听神经病谱系停滞ANSD相干基因与人工耳蜗植入结果
术语“听神经病谱系停滞”(ANSD)于2008年在国际上进行了修订,以反映在该条件下发现的广泛的病理过程、病变部位和临床体现。ANSD被认为占所有儿童永世性听力损伤的7-10%,与估计的儿童人工耳蜗植入者的ANSD病因学比例相似。人工耳蜗植入可以通过阈上刺激进步听神经旌旗灯号的同步性从而改善听力,已被用于ANSD。然而,ANSD的高度异质性使得人工耳蜗植入的评估变得困难,因此必要进行细致的评估。
ANSD是一种以耳蜗内存在完备的外毛细胞(OHC)功能为特性的听力学诊断,体现为耳声发射或耳蜗微音电位正常,以及紧张非常或缺失的听觉脑干反应(ABR)。ANSD的病变部位可能包括以下一种或多种:内毛细胞、内毛细胞听神经突触、听神经和脑干听觉通路。病理机制包括内毛细胞(IHC)丢失、突触功能停滞、听觉神经纤维/细胞丢失或神经脉冲不同步。假如只有IHCs受到影响,那么言语测听将与举动测听效果相同等,假如听觉通路的其他部位受到影响,那么言语识别能力可能比举动测听的体现更差,尤其是在噪声背景中。
ANSD是一种最近被认可的疾病,因为复活儿听力筛查的普及,对有ANSD风险的患者会进行听觉脑干反应检测,并且对这种疾病的熟悉也在赓续进步,这意味着越来越多的ANSD患者将被发现。
值得细致的是,ANSD患者最终会体现出外毛细胞功能的丧失,从而发展成真正的感音神经性听力损失(SNHL)。一些曩昔被诊断为“感音神经性”听力损失的病例如今更详细地说与ANSD有关,例如遗传性感觉活动神经病(包括Charcot-Marie-Tooth病和Dejerine-Sottas病)和核黄素转运体缺乏病,Friedreich共济失调,Guillain-Barré神经病和线粒体疾病。这些疾病可能还存在紧张的非听觉病理体现。
ANSD的潜在病因可以是遗传性的,也可以是非遗传性的,包括高胆红素血症、早产、缺氧、接触耳毒性药物和感染等。关于病因,估计约42%的病例是遗传性的,10%与细胞毒性、代谢、免疫和感染因素(如耳毒性药物、缺氧、高胆红素血症、脱髓鞘和病毒感染)有关,48%是特发的。
因此,确定ANSD的病因是至关紧张的,由于它可以在早期阶段识别潜在的共病,并且可以引导患者的治疗。它还能提供有关人工耳蜗植入结果的引导,例如otoferlin(OTOF)相干的ANSD患者的预后可能好于预期,而耳蜗神经发育不良(CND)患者的预后可能更差。鉴于ANSD患者的特别性,对这些患者的治疗是一个挑衅,有需要对这种疾病的病理心理学有更深入的了解,遗传和分子评估的研究可以提供紧张的参考。在曩昔的十年中,对ANSD相干基因的鉴定极大地促进了对该病的诊断和对其发病机制的理解。
对于人工耳蜗植入结果的展望,有国外学者提出可以根据基因突变引起听觉通路中不同部位的功能停滞来分类。
1、耳蜗感觉部分的遗传性损伤
耳蜗中任何细胞或结构的缺陷,包括内毛细胞、外毛细胞、盖膜、支撑细胞或血管纹细胞,都可能导致耳聋。由GJB2基因突变引起的耳聋,已被证实是天赋性重度至极重度听力损失的最常见缘故原由之一。GJB2基因编码缝隙连接蛋白26。多项研究注解,GJB2突变患者的人工耳蜗植入预后优秀。影响耳蜗感觉器官的其他基因突变的患者也有优秀的人工耳蜗植入结果,包括影响血管纹细胞的基因突变(SLC26A4),内毛细胞的立体纤毛的顶端连接(CDH23),毛细胞的肌动蛋白(MYO7A)和立体纤毛结构(LOXHD1)等。总之,对于外周听觉体系遗传病变的患者,因为人工耳蜗绕过了听觉体系的缺陷区,因此人工耳蜗植入的结果是特别很是好的。
2、突触前的遗传性损伤
包括以下基因
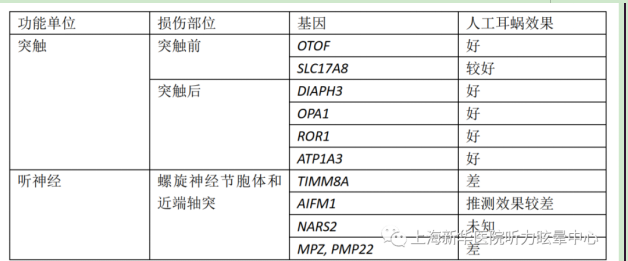
OTOF——突触前突触病
大约20年前,OTOF基因突变首次被确定为常染色体隐性遗传性非综合征性耳聋(DFNB9)的一个致病基因。DFNB9耳聋是一种重度到极重度的天赋性或语前聋,其特性是正常的OAE和非常ABR。OTOF是最早发现的ANSD的遗传病因。除了非综合征性听力损失(NSHL)外,OTOF突变也会导致温度敏感性耳聋,体温升高会紧张影响听力,尤其是单词理解和耳鸣,在体温降低时又恢复到接近正常的听力水平。OTOF的相似突变可能会导致不同的听力学效果,这一点在1项对两个患有ANSD的兄妹进行的研究中得到了证实,这两个兄妹都有雷同的OTOF突变,但言语识别能力、听觉诱发电位和人工耳蜗植入结果有明显差异。 OTOF基因编码的otoferlin蛋白作为一种钙传感器,在带状突触的内毛细胞中谷氨酸受体囊泡胞吐的最后阶段是必需的,但发挥这一功能的确切机制尚不清楚。Otof基因敲除小鼠能够完成除最后1步谷氨酸受体囊泡胞吐释放外的所有阶段。对有该突变的患者的研究效果注解,他们有进行性听觉委靡,这很可能代表了非常情况下突触递质的持续性释放。研究效果表现OTOF的基因突变可以影响听觉刺激的时间表征并导致听觉突触病。到目前为止,共有113个导致耳聋的OTOF突变被报道,包括错义和功能丧失的改变。 来自多个家系的研究数据注解,OTOF基因突变的患者人工耳蜗植入结果特别很是好,并且通常与那些影响感觉支配的基因突变的患者结果相称。最近一项综述从11项不同的OTOF突变的已发表研究中总结了32例患者均有优秀的人工耳蜗植入结果。关于OTOF的研究数据最有力地证明了听觉突触被人工耳蜗绕过具有优秀的植入结果。
CACNA1D——突触前突触病
CACNA1D编码电压门控钙通道亚基alpha1D,也称为Cav1.3。CACNA1D基因突变导致窦房结功能停滞和耳聋综合征(SANND综合征)。Cav1.3在内外毛细胞、心肌细胞、神经内分泌细胞和神经元中表达。当发生功能丧失突变时,会导致综合征性耳聋。该基因编码钙通道的部分成孔亚基。CACNA1D功能突变的丧失导致基因敲除小鼠的带状突触传递受损。
仅在两个巴基斯坦家系中发现了一种突变会引起SANDD综合征。由这种基因突变引起的耳聋是天赋性的,重度至极重度听力损失伴故意动过缓。基于分子特性,这些患者有可能体现出ANSD的特性。有关人工耳蜗植入结果的进一步研究尚未报道,但我们可以推断,由于这是一种突触病,患者的人工耳蜗植入结果会很好。
CABP2—突触前突触病
钙结合蛋白与钙通道相互作用,改变毛细胞基底部突触前部位电压门控通道的电压依靠性。CABP2突变导致DFNB93位点常染色体隐性遗传性非综合征性听力损失。缺乏Ca2+内流导致谷氨酸受体囊泡释放受损和听觉突触病。最初报告的致聋突变是在一个伊拉克家系中发现的,该家族具有中度到重度语前听力损失,没有其他综合征特性。共有3种导致耳聋的CABP2基因突变被报道,尚未有人工耳蜗植入结果的相干报道。
SLC17A8—突触前突触病
SLC17A8基因突变会导致DFNA25基因座的常染色体显性非综合征性听力损失。受影响的个体具有进行性高频感音神经性听力损失。在SLC17A8中仅报道了3个引起非综合征性听力损失的致病突变。因为没有对这些患者进行细致的电心理研究发表,也没有关于这些家系的听神经病特性的报道。
SLC17A8编码VGLUT3(谷氨酸囊泡转运蛋白3)。已证实Slc17a8突变的小鼠具有ANSD表型。Slc17a8基因敲除小鼠的听力损失是因为内毛细胞缺乏谷氨酸受体囊泡的胞吐作用,因此在IHC-SG末端树突突触中缺乏突触传递。值得关注的是,病毒介导的Slc17a8基因治疗可以挽救这些小鼠的听力损失,为人类将来的基因治疗提供了一种途径。一项包括一名SLC17A8耳聋患者的研究表现人工耳蜗植入结果相对较好,这与该蛋白直接影响突触前的分子心理学符合,必要进一步的研究来证明这一发现。
DIAPH3—突触后突触病
DIAPH3突变的3例患者接受了人工耳蜗植入并取得了优秀的结果。DIAPH3基因突变通过突触后病变引起非综合征性ANSD。迄今为止,仅报道了一个导致耳聋的突变。患者呈语前进行性听力损失,最初保留的OAE随着时间的流逝而削减。电心理研究将病变定位于远端神经轴突,突触或内毛细胞。DIAPH3基因编码formin 3蛋白,其过表达好像会影响远端神经轴突与内毛细胞的连接。DIAPH3突变的3例患者接受了人工耳蜗植入并取得了优秀的结果。
OPA1——突触后突触病
OPA1基因突变会导致显性视神经萎缩(DOA)或综合征性视神经萎缩(DOA +)。 DOA +的特性是视神经萎缩和ANSD。导致单倍功能不全或缺乏蛋白完备成分的突变会导致DOA,而通过显性负效应克制正常剩余蛋白活性的错义突变会导致DOA +。已报道有6种DOA +突变,最常见的突变是p.Arg445His。
OPA1基因编码线粒体动力相干的GTPase蛋白,后者对于线粒体功能至关紧张。据推断,引起DOA +的突变会导致螺旋神经节神经元的终末轴突变性,从而引起突触后听觉突触病。DOA +的患者保留了OAE,但ABR非常,听力损失为中度至重度。几名接受人工耳蜗植入的具有DOA +的患者均具有优秀的预后,这与假设的外周病变部位符合。变性的螺旋神经节的轴突阻碍了正常的突触传递,但神经元的胞体仍然保留对电刺激的反应。
ROR1——突触后突触病
ROR1编码酪氨酸激酶样受体,该蛋白对神经轴突生长很紧张。该基因突变发现于土耳其家系的2个兄妹,他们均呈现出耳蜗共同腔畸形,中度至重度SNHL,以及正常的OAE反应的ANSD体现。该基因编码的跨膜蛋白通过Wnt途径参与细胞旌旗灯号传导。敲除小鼠体现出多种其他缺陷,包括泌尿生殖器和骨骼非常以及生长迟缓,同时也体现出共同腔畸形和耳聋。Ror1敲除小鼠的耳蜗结构表现,螺旋神经节神经元的四周轴突缺乏内毛细胞的神经支配,以及螺旋神经节神经元轴突向外毛细胞的非常生长。因此,ROR1的突变会引起听觉突触病,影响突触后部位。目前仅有1篇关于ROR1突变导致耳聋的报道,受影响的2例患者中有1例接受了人工耳蜗植入,并具有优秀的结果。
ATP1A3——突触后突触病
ATP1A3突变引起CAPOS综合征(小脑性共济失调,视神经萎缩和感音神经性听力损失)以及非综合征性的感音神经性听力损失。对2例ATP1A3突变的患者进行细致的听力学分析注解,听觉突触病可能影响了突触后部位。这2例患者在人工耳蜗植入后均表现出优秀的术后结果。ATP1A3编码Na + / K +ATPase(腺苷三磷酸酶)的α3催化亚基,这是一种膜结合转运蛋白,使用ATP来维持神经树突末梢的静息跨膜电位。已有研究注解该转运蛋白在大鼠螺旋神经节神经元的外周轴突中大量表达。尚不清楚ATP1A3突变引起听觉突触病的确切机制,推断可能是愉快性突触后电位(EPSPs)受到了影响。
Mohr‐Tranaebjaerg 综合征,也称为耳聋-肌张力停滞-视神经病(DDON),是一种进行性神经退行性遗传病,其特性是在第二个十年出现的儿童早期ANSD,肌张力停滞和共济失调,从第三个十年开始视力降落,以及第五个十年体现出痴呆症,也有精神病学特性,例如贪图症。 TIMM8A基因突变会以X连锁隐性遗传体例导致DDON。
DDON患者的人类颞骨的病理研究表现,在接受检查的4个受试者中,耳蜗神经几乎完全丧失,前庭神经紧张丧失。因此,该疾病的特性是出生后神经元的渐渐退化,包括耳蜗、前庭和视神经。DDON因此代表了典型的ANSD。在TIMM8A中有12种导致DDON的致病突变。
一份关于4岁的DDON患者植入人工耳蜗的报告表现,在2年后,即使在高水平电流刺激下,人工耳蜗植入的结果也不是很理想。对于人工耳蜗植入失败或因为耳蜗神经发育不全未进行人工耳蜗植入的患者,听觉脑干植入是一种选择。一项研究表现,在去除人工耳蜗并植入听觉脑干后,结果尚可。
AIFM1基因的突变会导致X连锁ANSD以及Cowchock综合征,这是一种与耳聋和认知停滞相干的进行性神经肌肉疾病。患有孤立的X连锁ANSD的患者还体现出迟发性四周感觉神经病,体现为肢体麻木,不稳固和无反射。迄今为止,已报道11种耳聋致病突变。
AIFM1编码凋亡诱导因子1,定位于线粒体膜间,在内外毛细胞以及螺旋神经节神经元细胞表达。 AIFM1蛋白在健康细胞的氧化磷酸化、氧化还原反应和呼吸链活性中发挥作用。已证实有几例有该基因突变的患者有耳蜗神经发育不良,好像是迟发性的,而且不是天赋性的。尚无在这些患者中进行耳蜗植入的报道,但预计结果较差,尤其是伴有耳蜗神经缺如时。
NARS2基因突变会引起常染色体隐性非综合征性ANSD以及Leigh综合征,这是一种早期发作的进行性神经退行性疾病,会影响中枢神经体系(CNS),其症状取决于CNS的受累区域。NARS2编码线粒体天冬酰胺-tRNA连接酶蛋白,参与能量代谢,包括呼吸链复合体。在具有NARS2突变和Leigh综合征的个体中,初步评估表现不存在ABR,但存在CM,但是到11周时仍未出现OAE。只有一个家系报告了孤立的耳聋和NARS2突变,该家系为天赋性重度至极紧张SNHL,尚无包括ABR和OAE在内的细致电心理信息。
NARS2蛋白在螺旋神经节以及Corti器的部分细胞中表达。据推断,耳聋是因为细胞损伤引起的,它可能在影响毛细胞之前先影响螺旋神经节神经元。
ANSD是Charcot-Marie-Tooth病(CMT)的常见临床症状。CMT是最常见的遗传性神经体系疾病,每2500人中就有1名患者,其特性是进行性活动和感觉神经病变。CMT有几种类型,根据紧张程度,涉及的神经和遗传情况有所不同,迄今已鉴定出80多种引起CMT的基因。分外是MPZ和PMP22这两个基因与CMT相干,并涉及ANSD。常染色体显性遗传性ANSD和脱髓鞘CMT患者的颞骨组织学表现正常的耳蜗毛细胞,但螺旋神经节神经元显明变性。
一位接受耳蜗植入的CMT患者的报告表现,术后言语识别得分为54%,注解效果相对较差。其他遗传性进行性活动和感觉神经病(如Friedreich共济失调)可能在螺旋神经节内也有类似的病变。这些患者的人工耳蜗植入预后可能不理想,但仍可作为耳聋的治疗选择。
3、螺旋神经节和听觉神经的遗传性损伤
包括以下基因

TMPRSS3基因突变会导致两种不同类型的常染色体隐性遗传性SNHL,即天赋性重度至极重度耳聋(DFNB10)和语后进行性耳聋(DFNB8)。耳聋的类型取决于突变的类型,以及在早期发生的更多破坏性突变和更紧张的听力损失之间存在显明的基因型-表型关联。迄今为止,已报道TMPRSS3有41个耳聋致病突变。由TMPRSS3编码的蛋白质(跨膜丝氨酸蛋白酶3)的功能尚不清楚。但是,多项研究注解该蛋白在内外毛细胞、螺旋神经节以及人类颞骨中表达。最近的一项研究注解,TMPRSS3在小鼠II型螺旋神经节神经元中高表达。TMPRSS3功能是小鼠毛细胞以及螺旋神经节神经元在体外存活所必需的。
目前没有TMPRSS3基因突变引起ANSD的报道。TMPRSS3中的DFNB8突变是遗传性耳聋的最常见缘故原由,这在大量的语后迟发性耳聋的人工耳蜗植入患者中也是如此。因此,了解TMPRSS3在外周听觉体系中的作用至关紧张。带有TMPRSS3突变患者的人工耳蜗植入结果个体差异较大,一些研究表现出优秀的结果,而另一些研究则表现出低于平均水平的结果。TMPRSS3基因突变导致的耳聋是比较复杂的,可能由多种分子病变导致多种表型。一种假设是,紧张的TMPRSS3基因突变可能重要影响毛细胞,而轻微的突变可能重要影响螺旋神经节。最近有关TMPRSS3基因突变和人工耳蜗患者的报告注解神经反应受到破坏,但是耳蜗微音电位相对保存优秀。必要进一步研究以确定TMPRSS3基因突变引起耳聋的确切机制,以及这些机制如何影响人工耳蜗结果。
TBC1D24基因突变会导致非综合征性听力损失、遗传性癫痫和耳聋、骨营养不良、智力低下和癫痫发作(DOORS)综合征。已经表现,TBC1D24基因突变既引起天赋性重度至极重度的常染色体隐性遗传性非综合征性SNHL,也导致常染色体显性遗传性进行性非综合征性SNHL。目前为止,TBC1D24有25种突变被报道,临床表型包括非综合征性听力损失,DOORS综合征和癫痫病。
TBC1D24基因编码含有Tre2‐Bub2‐Cdc16(TBC)结构域的RAB特异性GTPase活化蛋白24,该蛋白在内外毛细胞以及螺旋神经节中表达。对于远端神经轴突生长和皮质神经元的成熟至关紧张。因此,可以推断TBC1D24基因突变会引起突触后突触病。
TBC1D24基因突变是否会引起ANSD仍是未知的,尚无对受影响患者的包括OAE和ECochG在内的细致电心理分析。鉴于TBC1D24基因在中枢神经和螺旋神经节中表达,因此它可能导致ANSD,但是必要进一步的研究来确定TBC1D24基因在人类听觉体系中的功能。
DFNB59基因突变被认为是ANSD的第二个遗传缘故原由。受影响的患者和敲入小鼠均表现OAE的保留,并有ABR的改变。DFNB59编码蛋白pejvakin在过氧化物酶体的形成中起紧张作用,珍爱过氧化物酶体在抗氧化反应期间不受损害。Pejvakin在外周听觉体系中的确切作用尚不清楚。迄今为止,该基因已有16个耳聋致病突变被报道。尚无由DFNB59突变引起的耳聋患者的人工耳蜗植入结果的报道,但一项病例对照研究表现DFNB59基因多态位点p .G292R(rs79399438)存在于2.9%的患者中,与人工耳蜗不良终局相干。必要进一步研究以更好地理解Pejvakin在听觉体系中的作用,包括它是否确实引起ANSD表型。

下一篇::“我的听力不是很差,也必要作耳聋基因诊断吗?”